You appear to be using incognito/private browsing mode or an ad blocker, which may adversely affect your experience on the site. Please disable any ad blockers and view the site in non-private mode.
KAPA Library Quantification Kits contain all the reagents needed for the accurate, reliable and reproducible qPCR-based quantification of next-generation sequencing (NGS) libraries prepared for sequencing on Illumina platforms. Kits include KAPA SYBR FAST qPCR Master Mix (formulated with different passive reference dyes for different qPCR instruments), a platform-specific library quantification primer premix, and a pre-diluted set of DNA standards. Refer to the Instrument Compatibility Chart for guidance on compatible platforms.
Benefits of KAPA Library Quantification Kits
Reliable and sensitive quantification of all sequencing-competent library molecules
Accurate and reproducible quantitation across a wide range of library types, concentrations, fragment length distributions and GC content
Accurate, equimolar pooling for multiplexed sequencing
Flexibility to support manual and automated high-throughput pipelines as well as PCR-free workflows
What makes KAPA Library Quantification Kits highly efficient for library quantification?
KAPA Library Quantification Kits contain KAPA SYBR FAST DNA Polymerase, which was engineered through our directed evolution technology to amplify diverse DNA fragments with similar efficiency. This is important for the amplification of heterogeneous populations such as NGS libraries. Library quantification kits containing wild-type DNA polymerases only count “easy” library molecules.*
View data showing efficient amplification and quantification of diverse libraries by KAPA SYBR FAST DNA Polymerase compared to other options.
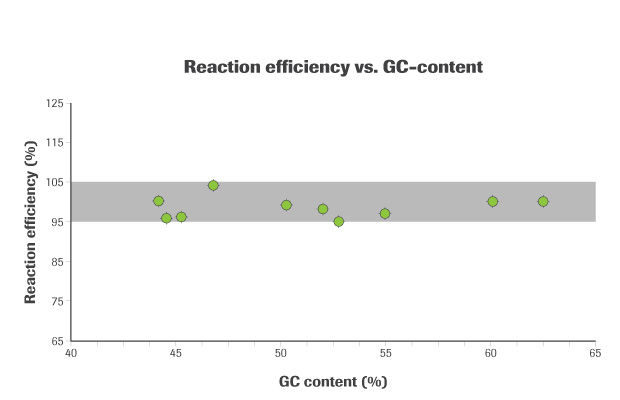
Figure 1. KAPA SYBR FAST exhibits high-efficiency amplification and library quantification. Amplification efficiencies achieved with KAPA SYBR FAST for ten diverse amplicons, plotted against GC content. All ten amplicons were amplified with similar efficiency (within the optimal range of 95% –105%). This confirms that KAPA SYBR FAST is the ideal enzyme for the amplification of heterogenous populations of DNA fragments, such as NGS libraries.
How do KAPA Library Quantification Kits Work?
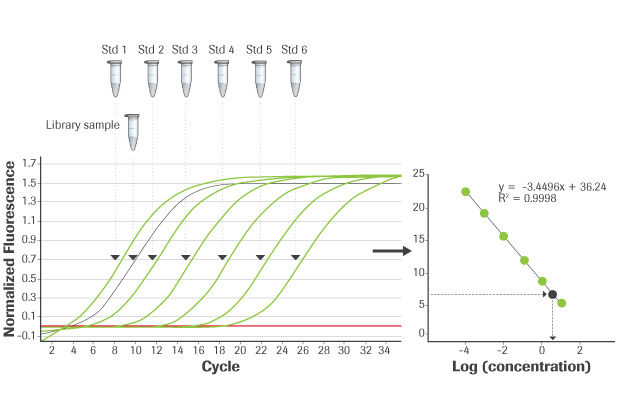
Generation of standard curve and quantification of library concentration:
Six pre-diluted DNA standards and appropriately diluted NGS libraries are amplified using platform-specific qPCR primers that target adapter sequences. The average Cq value for each DNA standard is plotted against its known concentration to generate a standard curve. The standard curve is used to convert the average Cq values for diluted libraries to concentration, from which the working concentration of each library is calculated.
Product highlights
The complete library solution
All reagents needed for absolute, qPCR-based quantification of individual NGS libraries or indexed library pools
Standard curves to support all library construction workflows, including PCR-free methods
Quantification of only/all sequencing-competent library fragments
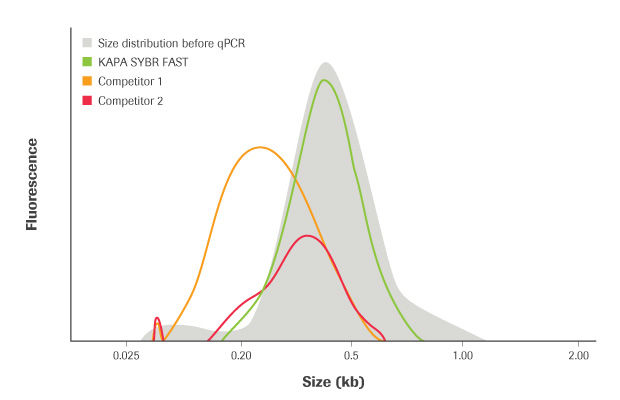
Figure 3. DNA fragment size distributions for an NGS library before (grey fill) and after qPCR amplification with KAPA SYBR FAST and two competitor kits containing wild-type Taq polymerase. Reactions were performed with the following cycling protocol: 95°C for 10 min, followed by 40 cycles of 95°C for 10 sec, and 60°C for 45 sec. The KAPA SYBR FAST quantification product is representative of the template DNA (NGS library), whereas the quantification products generated with wild-type enzymes are not.
High consistency and product quality with a proven track record
Use of single DNA standard for all library types
Pre-diluted standards with very high lot-to-lot consistency
View data showing high consistency in quantification across library types and consistent amplification efficiency by KAPA Library Quantification Kits.
Figure 4. High consistency across different library types and across hundreds of production lots. Nine human WGS libraries (41% GC) and two microbial WGS libraries (Rhodococcus sp., 70% GC and Staphylococcus sp., 35% GC) were quantified with distinct lots (Lots 1 and 2), and distinct sets of reagents from the same lot (Set 1 and 2) of KAPA Library Quantification Kit for the Illumina platform.
Reproducible cluster density from samples of variable quality
Optimal and predictable cluster densities to maximize sequencing capacity and throughput
Reliable results for all library types, including from low-quality FFPE DNA
Equimolar pooling for multiplexed sequencing
Equimolar pooling of indexed libraries, irrespective of pre-pooling library concentrations
Uniform distribution of reads across all the libraries in a pool
View data showing uniform distribution of reads in multiplexed sequencing pools using KAPA Library Quantification Kits.
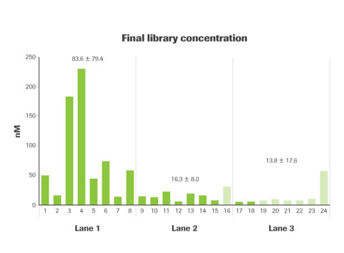
Figure 5. Accurate and sensitive quantification enables a
uniform distribution of reads in muliplexed sequencing pools.
Twenty-four indexed libraries, prepared from high-quality
human genomic DNA (darker bars) or FFPE DNA (lighter bars),
were quantified by qPCR using the KAPA Library Quantification
Kit for Illumina platforms and combined to create three
sequencing pools of equimolar concentration. Each pool was
sequenced in a different lane of the same flow cell on a HiSeq
2500 instrument. (Figure 5) The twenty-four individual libraries
represented a ~44-fold range of pre-pooling concentrations
(5.2 nM to 229.8 nM), whereas the sequencing read distribution
(Figure 6) only varied between 9.6% and 13.9%. The coefficient
of variation (CV) for pre-pooling library concentration was
94.9%, 49.4%, and 127.9% for the libraries pooled for
sequencing in lanes 1, 2, and 3, respectively. The KAPA Library
Quantification Kit enabled accurate equimolar pooling,
reducing the CV to 2.5%, 4.8%, and 11.2% after normalization.
Figure 6. Accurate and sensitive quantification enables a uniform distribution of reads in muliplexed sequencing pools. Twenty-four indexed libraries, prepared from high-quality human genomic DNA (darker bars) or FFPE DNA (lighter bars), were quantified by qPCR using the KAPA Library Quantification Kit for Illumina platforms and combined to create three sequencing pools of equimolar concentration. Each pool was sequenced in a different lane of the same flow cell on a HiSeq 2500 instrument. (Figure 5) The twenty-four individual libraries represented a ~44-fold range of pre-pooling concentrations (5.2 nM to 229.8 nM), whereas the sequencing read distribution (Figure 6) only varied between 9.6% and 13.9%. The coefficient of variation (CV) for pre-pooling library concentration was 94.9%, 49.4%, and 127.9% for the libraries pooled for sequencing in lanes 1, 2, and 3, respectively. The KAPA Library Quantification Kit enabled accurate equimolar pooling, reducing the CV to 2.5%, 4.8%, and 11.2% after normalization.
Complete kits include KAPA SYBR FAST qPCR Master Mix (2X), Primer Premix (10X) and a set of 6 DNA standards. Reagents for qPCR (KAPA SYBR FAST qPCR Master Mix and Primer Premix) and DNA standards (1–6) are also sold separately. Where noted, Master Mixes contain instrument-specific reference dyes, while the Universal kit includes ROX High and ROX Low (both 50X) separately.
Generation of standard curve and quantification of library concentration:
Six pre-diluted DNA standards and appropriately diluted NGS libraries are amplified using platform-specific qPCR primers that target adapter sequences. The average Cq value for each DNA standard is plotted against its known concentration to generate a standard curve. The standard curve is used to convert the average Cq values for diluted libraries to concentration, from which the working concentration of each library is calculated.
Why is quantitative PCR a better option than standard protocols for NGS library quantification?
Standard protocols for all three major commercial Next Generation Sequencing (NGS) platforms employ unreliable, laborious, and costly methods for quantifying library DNA molecules prior to clonal amplification of sequencing templates. Accurate quantification of bona fide PCR-competent sequencing templates is crucial for reliable clonal amplification via bridge PCR (bPCR; “cluster amplification”) or emulsion PCR (emPCR) – underestimation results in non-clonality and/or over-clustering, while overestimation leads to poor yields of clusters or template-carrying beads.
Most standard methods for quantifying NGS libraries have a number of important disadvantages. First, electrophoresis and spectrophotometry measure total nucleic acid concentrations, whereas optimal cluster density or template-to-bead ratio depends on the appropriate input concentration of PCR-amplifiable DNA templates. Since the proportion of amplifiable DNA molecules in a library may vary with each sample, expensive and time-consuming titrations are required. Second, these methods have low sensitivity, consuming nanograms of precious samples, or about 1000 times more molecules than are required for sequencing. Finally, electrophoresis and spectrophotometry are not suited to high-throughput of samples, requiring laborious and error-prone manual liquid handling.
Why is quantitative qPCR inherently well-suited for NGS library quantification?
Specifically quantifies PCR-competent DNA molecules
Accurate across a very broad dynamic range
Amenable to automated liquid handling
Cost-effective
Allows accurate quantification of very dilute libraries
Consumes small amounts of sample
Why are there differences between the concentration obtained using the qPCR-based KAPA Library Quantification Kit method and the Agilent Bioanalyzer/Invitrogen qubit/spectrophotometry-based assay?
We believe that qPCR quantification of libraries is the best approach for minimizing variability in cluster density or bead enrichment. Aside from errors in data collection, processing, and/or analysis there are three possible explanations for such differences:
qPCR “counts” only those library molecules that are competent templates for PCR, and is therefore blind to all library molecules that cannot give rise to clusters during the bridge PCR process of cluster amplification or beads carrying amplified sequencing template during emPCR. We have therefore found that qPCR usually provides a lower estimate of library concentration than the less specific methods such as Agilent Bioanalyzer or dye-assisted spectrophotometry, e.g Picogreen.
Pipetting accuracy is a common source of error in all library quantification methods, especially when small volumes are measured, and when serial dilutions are performed. Because qPCR is highly sensitive, requires only tiny amounts of sample, and utilizes low reaction volumes (all advantages in other respects), it is particularly important to pay close attention to the quality and condition of the equipment used.
Electrophoresis accuracy: the stated coefficient of variation for quantification using the relevant Bioanalyzer DNA assays is 20%. Moreover, in our experience, Bioanalyzer assays are prone to erratic behavior depending on instrument maintenance, reagent age and storage conditions, operator error, etc. Other factors that may affect quantification by Bioanalyzer relate to the size distribution of library fragments. It is possible that the size cut-offs used to define the peak to be quantified might exclude fragments, especially at the smaller end, which would give rise to clusters or to enriched beads. Similarly, a relatively small change in the position of the calculated baseline, and/or the detection limit of the instrument/assay, may under- or over-estimate a significant number of library fragments at both ends of the size distribution.
How many libraries can I quantify with a single kit?
A single KAPA Library Quantification Kit is sufficient for the quantification of ~30 libraries (following our recommended protocols in a 96-well format). However, more libraries can be quantified using:
a 384-well plate format,
smaller reaction volumes,
fewer number of dilutions, and/or
fewer replicates.
Many variables will greatly affect the ultimate number of libraries that can be quantified per kit. Below are a few guidelines based on the details of our recommended protocols:
Many variables will greatly affect the ultimate number of libraries that can be quantified per kit. Below are a few guidelines based on the details of our recommended protocols:
Only one (preferably triplicate) set of six DNA standards needs to be run per assay.
Replicates*
Dilutions/library**
# Libraries/kit: 96 well***
# Libraries/kit: 384 well****
Reliability of Data
3
3
36
94
Very High
3
2
59
143
High
3
1
118
287
Acceptable, with increased risk of repeats
2
3
63
145
High
2
2
95
218
Acceptable, with increased risk of repeats
2
1
190
437
Acceptable, with increased risk of repeats
*We recommend triplicate reactions for the DNA standards and for the library samples; however, some users may feel sufficiently confident, especially after some experience with the kit, to do away with some replicates of the standards and/or the samples. This would make it difficult or impossible to troubleshoot any unexpected results and the quantification accuracy may suffer; on the other hand, many more libraries could be quantified with the same amount of reagent.
**We recommend that you perform three serial dilutions of each library sample to ensure that at least one dilution will fall within the upper limit of the range of the assay. This also allows calculation of the qPCR efficiency for that particular library sample. Thus, it is possible that with experience and/or with very standardized workflows, you may feel confident to stop doing these serial dilutions, in which case the number of qPCR reactions required for each quantification will be reduced.
***If you quantify a single library sample at a time, you will be able to quantify six libraries per kit of DNA standards. Each quantification experiment will use up 6 x 3 qPCR reactions for the standards plus 4 x 3 qPCR reactions for the library sample = 30 qPCR reactions in total. Since each Kit contains 5 mL KAPA SYBR FAST 2X qPCR Master Mix (sufficient for 500 x 20 μL reactions), you will have 500 μL – (6 x 30 μL) = 320 qPCR reactions left over when you have finished the DNA standards. In this case, additional DNA standards can be purchased separately, so that the leftover qPCR reagent is not wasted.
****Users may choose to run 10 μL volumes (especially in 384-well formats). The volume of DNA standard added to each reaction remains 4 μL, regardless of the reaction volume.
What is the primary reason for error and variability across the data points?
Inaccurate liquid handling is the most common reason for variability. Accurate quantification requires careful pipetting. For liquid handling systems, consult the relevant user manual. Alternatively, apply the following when using a non-automated device:
Please ensure that all reagents are completely thawed and thoroughly mixed before assembling your qPCR reactions.
After thawing and mixing, briefly centrifuge reagents to prevent droplets on tube walls from transferring to the outside of the pipette tip.
Concentrated solutions of DNA are viscous making it difficult to dispense small volumes. Ensure libraries and controls are thoroughly mixed before pipetting.
Use the recommended dilution buffer (10mM Tris-HCl, pH 8.0 (25˚C) + 0.05% Tween-20 when diluting libraries and controls. Tween-20 is a wetting agent and improves pipetting accuracy. Never dilute libraries in water, as DNA is sensitive to hydrolysis at pH>7.
Examine the tip before dispensing to ensure that the correct volume is being added.
Flush/rinse the tip 2–3 times after dispensing.
Use a new pipette tip every time.
Try to avoid placing the pipette tip too far under the surface when aspirating, as this may result in additional liquid adhering to the outside of the tip.
Dispense directly into the bottom of the tube or well.
Ensure that no residual liquid remains in the tip after dispensing.
Contamination of standards and libraries can also lead to error in the assay. We recommend:
good laboratory practice to avoid contamination of reagents, samples, consumables, pipettes and other equipment; and
a systematic decrease in the spacing of amplification curves for consecutive standards can indicate contamination with exogenous template or contamination of dilute DNA standard with more concentrated standards. To avoid the latter, dispense the DNA standards from the lowest to highest concentration (DNA standard 6 to DNA standard 1) and use fresh tips for each standard.
Why is the Cq of DNA standard 1 (and/or some of my library samples) too low (amplification is too early)?
Due to the design specifications and broad dynamic range of the KAPA Library Quantification assay, DNA standard 1 has a higher copy number than samples that are typically analyzed by a qPCR, particularly in relative quantification assays. As a result, standard 1 (and any samples that were not sufficiently diluted) may return a very early Cq score and/or an increasing fluorescent signal during the automated baseline determination performed by the qPCR instrument. Many instruments use the early cycles to calculate and set the baseline subtraction/correction, and samples that are changing significantly in fluorescence during this phase can dramatically affect this process (since there is no stable baseline).
It is usually possible to tell whether this is an issue by examining the amplification plots. If the corrected/processed amplification plot begins well below the baseline of samples crossing the Cq later, then it is likely that baseline determination/subtraction was not successful for that plot. Another useful way to judge whether an early Cq value has created problems during data analysis is to confirm that the expected spacing between consecutive standards is ~3.32 cycles (the standards represent a 10-fold dilution series). Similarly, consecutive two-fold dilutions of a given sample should cross the Cq ~1 cycle apart.
If you feel that the first standard and/or some sample dilutions have Cq scores that are too early, then simply exclude that standard and those sample dilutions from the analysis. This is most easily done by de-selecting them in the qPCR analysis software. If you experience this issue regularly with your instrument/settings/workflow, then you may choose to omit the first DNA standard from all future assays, and you might want to consider implementing a larger up-front dilution of your library samples in your standard workflow. If the first standard is omitted, ensure that the library Cq score falls within the dynamic range of standards 2-6.
What guidelines should i use to examine my standard curve?
When examining the data from the DNA standards, one should look at two things first:
The spacing between consecutive standards: The average ΔCq value between each pair of DNA standards (e.g. standard 1 and 2, or standard 2 and 3) is in the range of 3.1 to 3.6. If ΔCq is consistently outside of the range, this may indicate problems either with pipetting accuracy or problems with the efficiency of the qPCR reaction.
The calculated reaction efficiency is in the range of 90-110% and the R2 ≥ 0.99. If the assay returns a reaction efficiency that is within the acceptable range, but there is more than a 3% change in the value routinely achieved on the same qPCR instrument, this may indicate unexpected equipment variation and should be considered before accepting results.
The differences between the replicates: A difference of 1 Cq is equivalent to a 2-fold difference in template concentration. If the Cq score for any replicate varies by more than +/- 0.25 from the other two values, it should be discarded. The 0.25 is instrument dependent and may be determined for your particular instrument. More variance than this may indicate a large degree of well-to-well variation in the instrument (optics and/or temperature control), or problems with pipetting accuracy.
While it is acceptable to discard the occasional “outlier”, a large number of “outliers” generally indicates a problem either with the qPCR instrument or with the liquid handling equipment and/or technique. Further, the Cq values will vary significantly based on the qPCR instrument and threshold setting used.
Why does the recomended cycling protocol in the KAPA Library Quantification Kit protocol differ from that in the KAPA SYBR FAST qPCR Kit protocol?
Next-generation sequencing libraries are generally complex, comprising a very wide diversity of DNA fragments. While KAPA SYBR FAST qPCR reagents are generally capable of extremely fast thermocycling, we conservatively recommend relatively long denaturation and annealing/extension times for library quantification in order to accommodate the diversity of templates in a typical library sample. The recommended qPCR protocol for library quantification consists of an initial denaturation step at 95°C for 5 minutes followed by 35 cycles of denaturation at 95°C for 30 seconds and combined annealing/extension at 60°C for 45 sec. If the average library fragment size is >700 bp, increase annealing/extension time to 90 seconds.
Can melt curve analysis after qPCR library quantification be used to monitor the quality of the libraries (i.e detect adaptor dimers; average fragment sizes; library complexity; etc.)?
A melt curve analysis is typically included at the end of SYBR Green I-based qPCR assays to provide information about the specificity of the reaction. In library quantification melt curve analysis is less relevant, as the template being amplified is not comprised of a single DNA fragment of defined length and GC content, but is a heterogenous population of molecules, with a range of lengths and GC contents. Unlike the melt curve for a single amplicon, the melt curve for an Illumina library is a “composite” analysis, representing an average across the entire population of library fragments. It is also important to keep in mind that library samples are expected to reach their quantification cycle (Cq) relatively early, whereas the melt curve analysis is only performed after 35 cycles, i.e. at a time when substrate depletion has occurred and amplification artifacts that are no longer representative of the original template have been generated. This complicates the interpretation of melt curves generated in library quantification assays.
Nevertheless, we have found melt curve analysis useful for the identification of carry-over adapter dimer in Illumina libraries. Adapter dimers, formed during library construction with full-length adapters, are very efficiently amplified in the KAPA Library Quantification assay, and will lead to an overestimation of library concentration if present in significant quantities. A small second peak of higher temperature to the right of the main library peak will indicate adapter dimer. Please note that melt curves for the DNA standard included in KAPA Library Quantification Kits for Illumina platforms display a very characteristic double peak. This is the result of differential local melting in the 452 bp linear standard.
What are the primer sequences used in the various KAPA Library Quantification Kits?
Illumina
Primer P1: 5′-AAT GAT ACG GCG ACC ACC GA-3′
Primer P2: 5′-CAA GCA GAA GAC GGC ATA CGA-3′
How are the DNA standards in the KAPA library Quantification Kits quality controlled to minimize batch-to-batch variance and ensure reliability?
The DNA standards used in our Library Quantification Kits are not sequencing libraries, but rather a defined, pure, linear, dsDNA amplicon for each set of DNA standards. This allows us to rigorously validate their efficiency and reproducibility for use as qPCR amplification standards. Before accepting a newly manufactured lot into our inventory, we use a stringent qPCR assay to compare each new lot of KAPA Library Quantification DNA standards to a reference set of standards during manufacturing and quality control. We compare Cq scores for each standard in a newly manufactured set with Cq scores in a reference set of standards, and we ensure that each standard lies within 0.1 Cq of the respective reference standard, and that the resulting standard curve essentially lies on top of the reference standard curve (minimal deviations in y-intercepts and slopes).
What are the storage recommendations for KAPA Library Quantification Kits?
KAPA Library Quantification Kits are shipped on dry ice or ice packs, depending on the country of destination. Upon receipt, store the entire kit at -20°C in a constant-temperature freezer. When stored under these conditions and handled correctly, all kit components will retain full activity for at least six months from the date of receipt.
All components of the KAPA Library Quantification Kits – as well as the combined KAPA SYBR FAST/Primer Premix solution – are stable through more than 30 freeze/thaw cycles. We therefore recommend that all reagents are stored in the dark at -20°C when not in use. Nevertheless, these reagents are stable in the dark at 4°C for at least one week, and may be stored in this state for short-term use, provided that they are not contaminated with microbes and/or nucleases.
What volume should I add ROX low/high to my assay?
For certain real-time cyclers, the presence of ROX reference dye compensates for non-PCR-related variations in fluorescence detection. The fluorescence level of ROX reference dye does not change significantly during the course of real-time PCR, but provides a stable baseline against which PCR-related fluorescent signals are normalized. Thus, ROX dye compensates for differences in fluorescence detection between wells due to slight variations in reaction volume or differences in well position. The presence of ROX dye in the master mix does not interfere with real-time PCR on any instrument, since the dye is not involved in the reaction and has an emission spectrum different from that of SYBR Green I. If using the kit with Universal qPCR Master Mix and will always only use ROX High or ROX low, the entire 200 µL of appropriate ROX solution may be added to the qPCR Master Mix when opened. If ROX will be added individually, add 0.4 μL of 50X ROX High/Low to each assay.